Pseudotime Spatial Trajectory Inference with Xenium Data¶
We will go through the tutorial on how to use stLearn to perform clustering (with or without imputation) and spatial trajectories inference with Xenium data from 10X Genomics. The main dataset we are working on is the breast cancer tumor microenvironment. You can download it here: https://www.10xgenomics.com/products/xenium-in-situ/preview-dataset-human-breast
1. Loading library¶
[1]:
import stlearn as st
import numpy as np
import matplotlib.pyplot as plt
st.settings.set_figure_params(dpi=120)
# Ignore all warnings
import warnings
warnings.filterwarnings("ignore")
/Users/andrew/conda/stlearn/lib/python3.10/site-packages/louvain/__init__.py:54: UserWarning: pkg_resources is deprecated as an API. See https://setuptools.pypa.io/en/latest/pkg_resources.html. The pkg_resources package is slated for removal as early as 2025-11-30. Refrain from using this package or pin to Setuptools<81.
from pkg_resources import get_distribution, DistributionNotFound
/Users/andrew/conda/stlearn/lib/python3.10/site-packages/stlearn/tl/cci/het.py:206: NumbaDeprecationWarning: The keyword argument 'nopython=False' was supplied. From Numba 0.59.0 the default is being changed to True and use of 'nopython=False' will raise a warning as the argument will have no effect. See https://numba.readthedocs.io/en/stable/reference/deprecation.html#deprecation-of-object-mode-fall-back-behaviour-when-using-jit for details.
@jit(parallel=True, nopython=False)
[2]:
library_id = "Xenium_FFPE_Human_Breast_Cancer_Rep1"
[3]:
st.datasets.xenium_sge(library_id=library_id, include_hires_tiff=True)
2. Reading Xenium data¶
You can download all the data in the 10X Genomics resource webpage. For H&E image, because it was generated separately with the main xenium data, then you need to perform the registration by your self. Here we provide an example of a H&E image which is performed a manual registration by Dr. Soo Hee Lee. You can download it here: Link
{kind=link}
[4]:
data_dir = st.settings.datasetdir / "Xenium_FFPE_Human_Breast_Cancer_Rep1"
adata = st.ReadXenium(feature_cell_matrix_file=data_dir / "cell_feature_matrix.h5",
cell_summary_file=data_dir / "cells.csv.gz",
library_id=library_id,
image_path=data_dir / "he_image.ome.tif",
scale=1,
spot_diameter_fullres=15,
alignment_matrix_file=data_dir / "he_imagealignment.csv",
experiment_xenium_file=data_dir / "experiment.xenium",
)
Added tissue image to the object!
[5]:
adata
[5]:
AnnData object with n_obs × n_vars = 167780 × 313
obs: 'imagecol', 'imagerow'
var: 'gene_ids', 'feature_types', 'genome'
uns: 'spatial'
obsm: 'spatial'
3. Preprocessing¶
Here, we use scanpy to perform several steps of preprocessing
[6]:
# Filter genes and cells with at least 10 counts
st.pp.filter_genes(adata, min_counts=10)
st.pp.filter_cells(adata, min_counts=10)
[7]:
# Store the raw data for using PSTS
adata.raw = adata
[8]:
# Normalization data
st.pp.normalize_total(adata)
Normalization step is finished in adata.X
After normalization, we have two options. One is using log transformation or square root transformation (recommend by the 10X team)
[9]:
# Squareroot normalize transcript counts. We need to deal with sparse matrix of .X
adata.X = np.sqrt(adata.X.toarray()) + np.sqrt(adata.X.toarray() + 1)
# If the matrix is small, we don't need to convert to numpy array. I believe, Xenium data is always large
# adata.X = csr_array(np.sqrt(adata.X) + np.sqrt(adata.X + 1))
# If you want to use log transformation, please use:
# st.pp.log1p(adata)
[10]:
# Run PCA
st.em.run_pca(adata, n_comps=50, random_state=0)
PCA is done! Generated in adata.obsm['X_pca'], adata.uns['pca'] and adata.varm['PCs']
Apply stSME (optional)¶
stSME is optional as an imputation function. Due to the scope of this tutorial, we will not use it here, but we recommend you run if you have an H&E image in your dataset
[11]:
# apply stSME to normalise log transformed data
# st.spatial.SME.SME_normalize(adata, use_data="raw")
# adata.X = adata.obsm['raw_SME_normalized']
# st.pp.scale(adata)
# st.em.run_pca(adata,n_comps=50)
[12]:
# Compute neighborhood graph of cells using the PCA representation
st.pp.neighbors(adata, n_neighbors=25, use_rep='X_pca', random_state=0)
OMP: Info #276: omp_set_nested routine deprecated, please use omp_set_max_active_levels instead.
Created k-Nearest-Neighbor graph in adata.uns['neighbors']
3. Clustering¶
Here we use louvain clustering. You also can use leiden clustering method with scanpy.tl.leiden.
[13]:
st.tl.clustering.louvain(adata, random_state=0)
Applying Louvain cluster ...
Louvain cluster is done! The labels are stored in adata.obs['louvain']
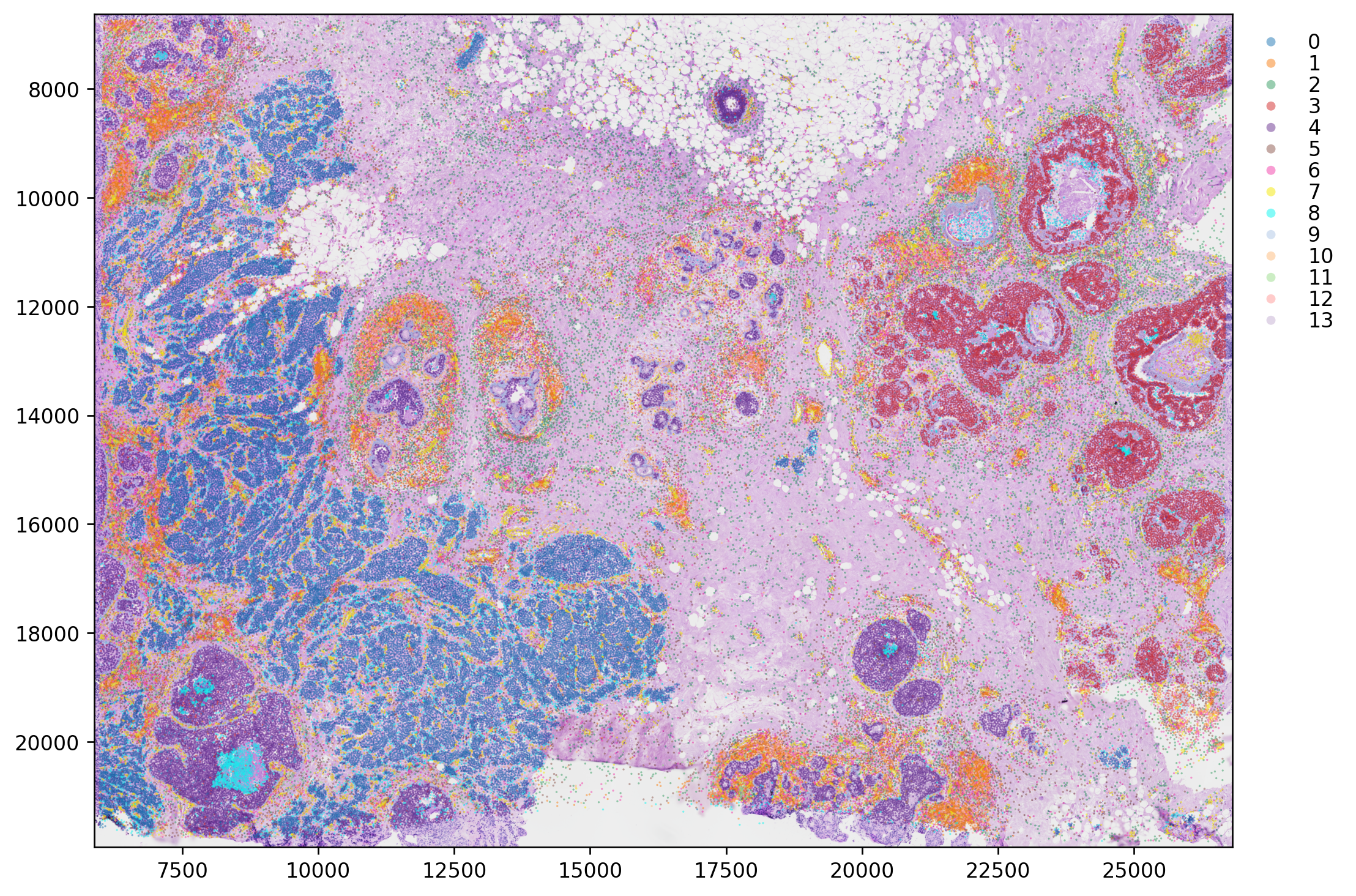
We can plot the clustering result:
[14]:
plt.rcParams["figure.figsize"] = (10, 10)
st.pl.cluster_plot(adata, use_label="louvain", image_alpha=1, size=1, cell_alpha=0.5, show_image=True, show_axis=True)
[14]:
AnnData object with n_obs × n_vars = 164000 × 313
obs: 'imagecol', 'imagerow', 'n_counts', 'louvain'
var: 'gene_ids', 'feature_types', 'genome', 'n_counts'
uns: 'spatial', 'pca', 'neighbors', 'louvain', 'louvain_colors'
obsm: 'spatial', 'X_pca'
varm: 'PCs'
obsp: 'distances', 'connectivities'
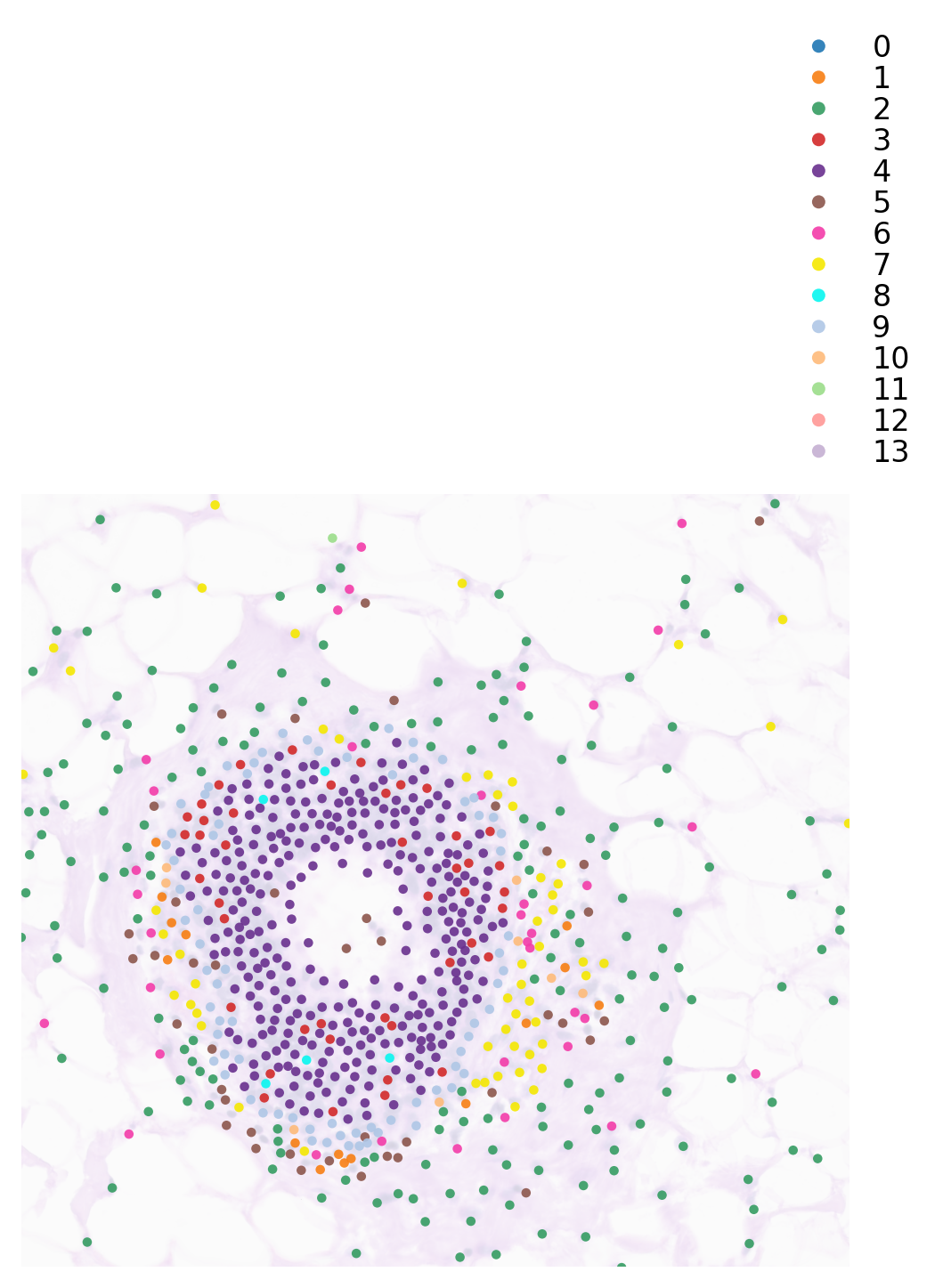
You can crop a region of interest with zoom_coord parameter.
[15]:
plt.rcParams["figure.figsize"] = (5, 5)
st.pl.cluster_plot(adata, use_label="louvain",
image_alpha=0.2, size=10,
cell_alpha=0.9,
zoom_coord=(17000.0, 18500.0, 7500.0, 8900.0),
show_color_bar=True)
[15]:
AnnData object with n_obs × n_vars = 164000 × 313
obs: 'imagecol', 'imagerow', 'n_counts', 'louvain'
var: 'gene_ids', 'feature_types', 'genome', 'n_counts'
uns: 'spatial', 'pca', 'neighbors', 'louvain', 'louvain_colors'
obsm: 'spatial', 'X_pca'
varm: 'PCs'
obsp: 'distances', 'connectivities'
4. PSeudo-Time-Space (PSTS) for spatial trajectory analysis¶
We provide PSTS for spatial trajectory analysis.
The first step is to select the root index. We allow the user to automatically select the index of the cell but still need to input which cluster should be the root cluster in your biological question.
In this tutorial, we focus on the cancer progression from Ductal carcinoma in situ (DCIS) to Invasive ductal carcinoma (IDC). That is why we choose the root cluster 6 as the DCIS cluster.
[16]:
adata.uns["iroot"] = st.spatial.trajectory.set_root(
adata,
use_label="louvain",
cluster="4",
use_raw=True
)
adata.uns["iroot"]
[16]:
14675
For the pseudotime function, we are based on the diffusion pseudotime algorithm to initialize the pseudotime for trajectory at the gene expression level. pseudotimespace_global is the function for incorporating the spatial information to construct the (meta) trajectory in the spatial context.
[17]:
st.spatial.trajectory.pseudotime(adata, eps=50, use_rep="X_pca", use_label="louvain")
All available trajectory paths are stored in adata.uns['available_paths'] with length < 4 nodes
In this dataset, the IDC cluster we are looking for is the one on the edge of ducts. This part is the first layer of invasive cancer cells starting from DCIS. That’s how we choose cluster 10 for the IDC.
[18]:
st.spatial.trajectory.pseudotimespace_global(adata, use_label="louvain", list_clusters=["4", "9"])
Start to construct the trajectory: 4 -> 9
[18]:
AnnData object with n_obs × n_vars = 164000 × 313
obs: 'imagecol', 'imagerow', 'n_counts', 'louvain', 'sub_cluster_labels', 'dpt_pseudotime'
var: 'gene_ids', 'feature_types', 'genome', 'n_counts'
uns: 'spatial', 'pca', 'neighbors', 'louvain', 'louvain_colors', 'iroot', 'louvain_index_dict', 'paga', 'louvain_sizes', 'diffmap_evals', 'threshold_spots', 'split_node', 'global_graph', 'centroid_dict', 'available_paths', 'PTS_graph'
obsm: 'spatial', 'X_pca', 'X_diffmap'
varm: 'PCs'
obsp: 'distances', 'connectivities'
After construct the spatial trajectories from DCIS to IDC cluster. We can observe multiple meta-trajectories with sub-clusters which generated by spatial feature. With tree_plot, you can show all of the detail sub-clusters ids.
[19]:
st.pl.trajectory.tree_plot(adata, figsize=(10, 20), ncols=5)

[19]:
AnnData object with n_obs × n_vars = 164000 × 313
obs: 'imagecol', 'imagerow', 'n_counts', 'louvain', 'sub_cluster_labels', 'dpt_pseudotime'
var: 'gene_ids', 'feature_types', 'genome', 'n_counts'
uns: 'spatial', 'pca', 'neighbors', 'louvain', 'louvain_colors', 'iroot', 'louvain_index_dict', 'paga', 'louvain_sizes', 'diffmap_evals', 'threshold_spots', 'split_node', 'global_graph', 'centroid_dict', 'available_paths', 'PTS_graph'
obsm: 'spatial', 'X_pca', 'X_diffmap'
varm: 'PCs'
obsp: 'distances', 'connectivities'
Now, we can crop and discover the transition genes of sub-clusters meta-trajectory
[20]:
st.pl.cluster_plot(
adata,
use_label="louvain",
show_trajectories=True,
list_clusters=["4", "9"],
show_subcluster=True,
zoom_coord=(17000.0, 18500.0, 7500.0, 8900.0),
show_node=False
)
[20]:
AnnData object with n_obs × n_vars = 164000 × 313
obs: 'imagecol', 'imagerow', 'n_counts', 'louvain', 'sub_cluster_labels', 'dpt_pseudotime'
var: 'gene_ids', 'feature_types', 'genome', 'n_counts'
uns: 'spatial', 'pca', 'neighbors', 'louvain', 'louvain_colors', 'iroot', 'louvain_index_dict', 'paga', 'louvain_sizes', 'diffmap_evals', 'threshold_spots', 'split_node', 'global_graph', 'centroid_dict', 'available_paths', 'PTS_graph'
obsm: 'spatial', 'X_pca', 'X_diffmap'
varm: 'PCs'
obsp: 'distances', 'connectivities'
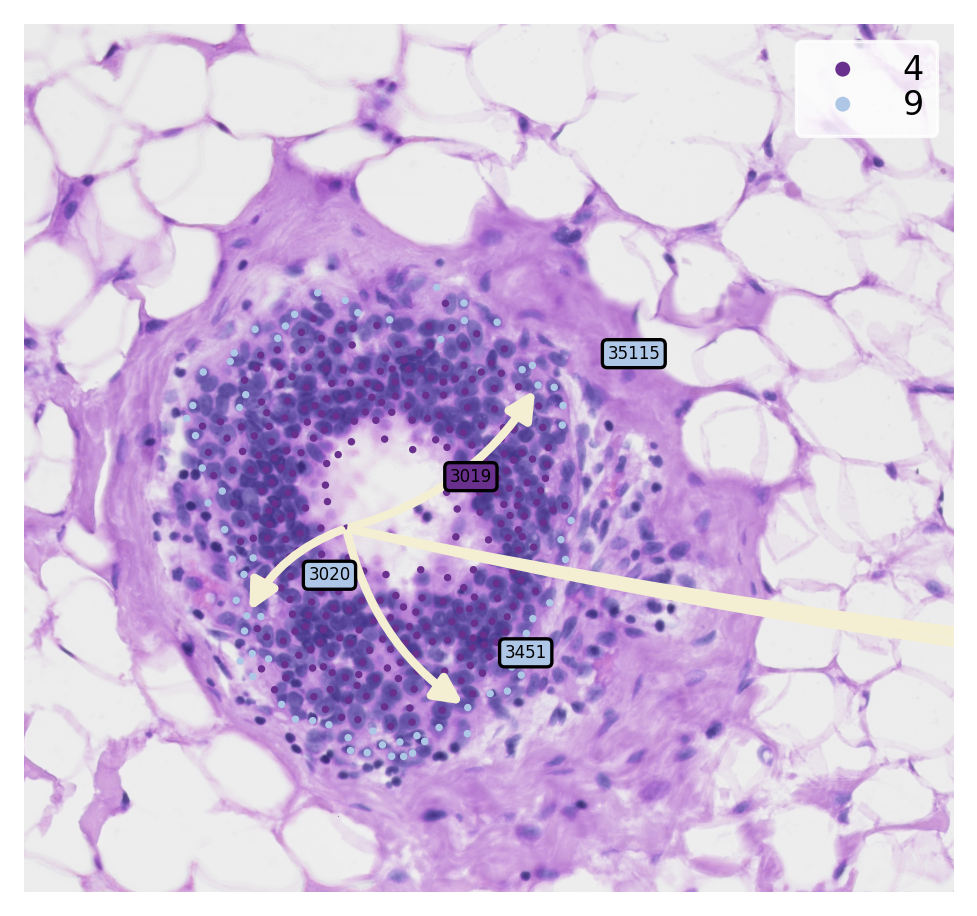
Finding the transition markers of clade_3019
[21]:
st.spatial.trajectory.detect_transition_markers_clades(adata, clade=3019, use_raw_count=False, cutoff_spearman=0.1)
Detecting the transition markers of clade_3019...
Transition markers result is stored in adata.uns['clade_3019']
Plot the transition markers, red color is down-regulated gene, blue color is up-regulated gene
[22]:
plt.rcParams["figure.figsize"] = (5, 5)
st.pl.trajectory.transition_markers_plot(adata, top_genes=20, trajectory="clade_3019")

[22]:
AnnData object with n_obs × n_vars = 164000 × 313
obs: 'imagecol', 'imagerow', 'n_counts', 'louvain', 'sub_cluster_labels', 'dpt_pseudotime'
var: 'gene_ids', 'feature_types', 'genome', 'n_counts'
uns: 'spatial', 'pca', 'neighbors', 'louvain', 'louvain_colors', 'iroot', 'louvain_index_dict', 'paga', 'louvain_sizes', 'diffmap_evals', 'threshold_spots', 'split_node', 'global_graph', 'centroid_dict', 'available_paths', 'PTS_graph', 'clade_3019'
obsm: 'spatial', 'X_pca', 'X_diffmap'
varm: 'PCs'
obsp: 'distances', 'connectivities'
We can observe some keratinocytes family genes here as the progression markers.
Acknowledgement¶
We would like to thank Soo Hee Lee and 10X Genomics team for their able support, contribution and feedback to this tutorial