stSME Comparison¶
This tutorial shows the stSME normalization effect between of two scenarios:
normal (without stSME)
stSME applied on raw gene counts
In this tutorial we use Mouse Brain (Coronal) Visium dataset from 10x genomics website.
[1]:
import scanpy as sc
import stlearn as st
import pathlib as pathlib
st.settings.set_figure_params(dpi=120)
# Ignore all warnings
import warnings
warnings.filterwarnings("ignore")
/Users/andrew/conda/stlearn/lib/python3.10/site-packages/louvain/__init__.py:54: UserWarning: pkg_resources is deprecated as an API. See https://setuptools.pypa.io/en/latest/pkg_resources.html. The pkg_resources package is slated for removal as early as 2025-11-30. Refrain from using this package or pin to Setuptools<81.
from pkg_resources import get_distribution, DistributionNotFound
/Users/andrew/conda/stlearn/lib/python3.10/site-packages/stlearn/tl/cci/het.py:206: NumbaDeprecationWarning: The keyword argument 'nopython=False' was supplied. From Numba 0.59.0 the default is being changed to True and use of 'nopython=False' will raise a warning as the argument will have no effect. See https://numba.readthedocs.io/en/stable/reference/deprecation.html#deprecation-of-object-mode-fall-back-behaviour-when-using-jit for details.
@jit(parallel=True, nopython=False)
[2]:
st.settings.datasetdir = pathlib.Path.cwd().parent / "data"
[3]:
data = sc.datasets.visium_sge(sample_id="V1_Adult_Mouse_Brain")
data = st.convert_scanpy(data)
[4]:
# pre-processing for gene count table
st.pp.filter_genes(data, min_cells=1)
st.pp.normalize_total(data)
st.pp.log1p(data)
Normalization step is finished in adata.X
Log transformation step is finished in adata.X
[5]:
# pre-processing for spot image
st.pp.tiling(data, out_path="tiling")
# this step uses deep learning model to extract high-level features from tile images
# may need few minutes to be completed
st.pp.extract_feature(data)
Tiling image: 100%|███████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████ [ time left: 00:00 ]
Extract feature: 100%|███████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████▉ [ time left: 00:00 ]
The morphology feature is added to adata.obsm['X_morphology']!
[6]:
# run PCA for gene expression data
st.em.run_pca(data, n_comps=50)
PCA is done! Generated in adata.obsm['X_pca'], adata.uns['pca'] and adata.varm['PCs']
OMP: Info #276: omp_set_nested routine deprecated, please use omp_set_max_active_levels instead.
(1) normal (without stSME)¶
[7]:
data_normal = data.copy()
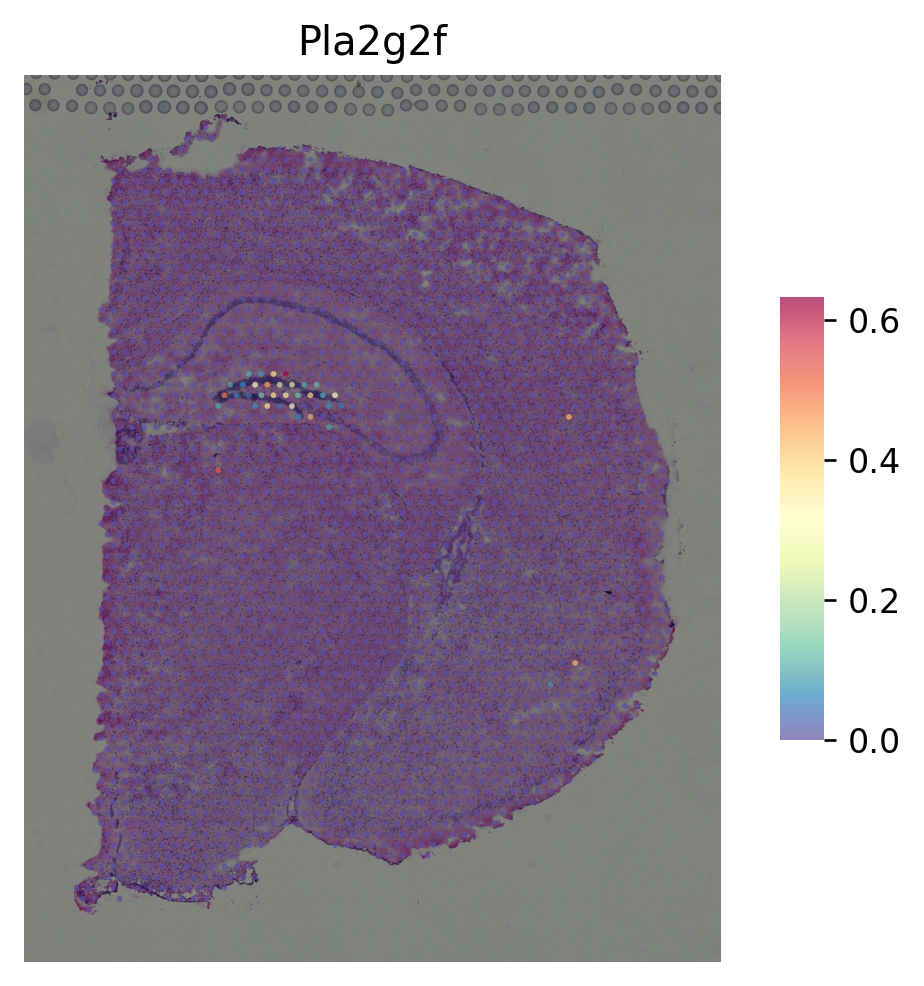
marker gene for CA3¶
[8]:
i = "Lhfpl1"
st.pl.gene_plot(data_normal, gene_symbols=i, size=3)
[8]:
AnnData object with n_obs × n_vars = 2702 × 21949
obs: 'in_tissue', 'array_row', 'array_col', 'imagecol', 'imagerow', 'tile_path'
var: 'gene_ids', 'feature_types', 'genome', 'n_cells'
uns: 'spatial', 'log1p', 'pca'
obsm: 'spatial', 'X_tile_feature', 'X_morphology', 'X_pca'
varm: 'PCs'
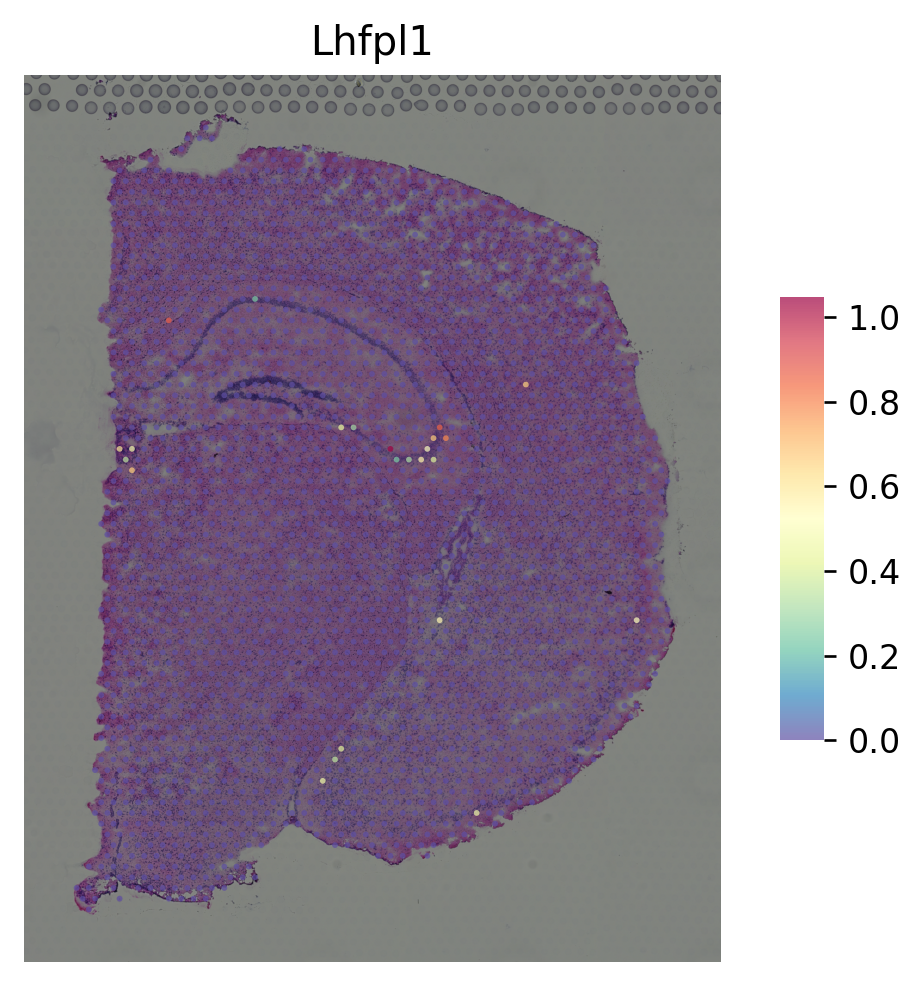
marker gene for DG¶
[9]:
i = "Pla2g2f"
st.pl.gene_plot(data_normal, gene_symbols=i, size=3)
[9]:
AnnData object with n_obs × n_vars = 2702 × 21949
obs: 'in_tissue', 'array_row', 'array_col', 'imagecol', 'imagerow', 'tile_path'
var: 'gene_ids', 'feature_types', 'genome', 'n_cells'
uns: 'spatial', 'log1p', 'pca'
obsm: 'spatial', 'X_tile_feature', 'X_morphology', 'X_pca'
varm: 'PCs'
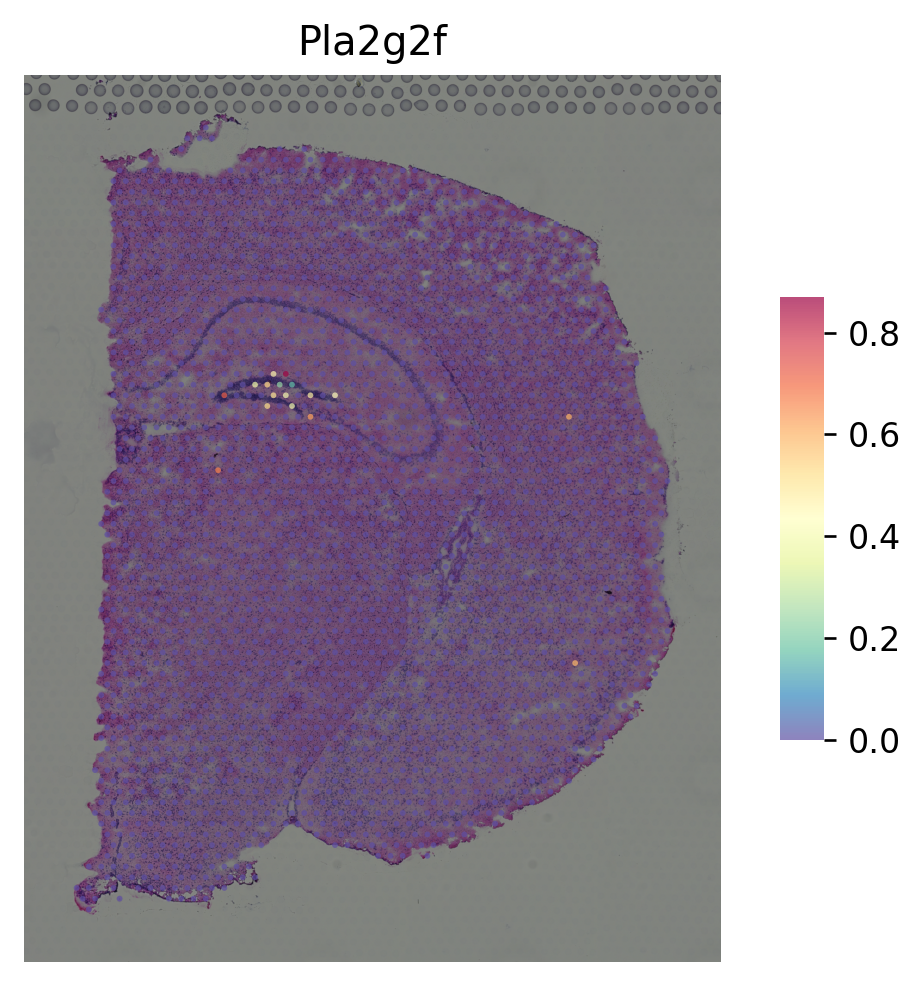
(2) stSME applied on raw gene counts¶
[10]:
data_SME = data.copy()
# apply stSME to normalise log transformed data
st.spatial.SME.SME_normalize(data_SME, use_data="raw")
data_SME.X = data_SME.obsm['raw_SME_normalized']
st.em.run_pca(data_SME, n_comps=50)
Adjusting data: 100%|█████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████ [ time left: 00:00 ]
The data adjusted by SME is added to adata.obsm['raw_SME_normalized']
PCA is done! Generated in adata.obsm['X_pca'], adata.uns['pca'] and adata.varm['PCs']
marker gene for CA3¶
[11]:
i = "Lhfpl1"
st.pl.gene_plot(data_SME, gene_symbols=i, size=3)
[11]:
AnnData object with n_obs × n_vars = 2702 × 21949
obs: 'in_tissue', 'array_row', 'array_col', 'imagecol', 'imagerow', 'tile_path'
var: 'gene_ids', 'feature_types', 'genome', 'n_cells'
uns: 'spatial', 'log1p', 'pca', 'gene_expression_correlation', 'physical_distance', 'morphological_distance', 'weights_matrix_all', 'weights_matrix_pd_gd', 'weights_matrix_pd_md', 'weights_matrix_gd_md'
obsm: 'spatial', 'X_tile_feature', 'X_morphology', 'X_pca', 'imputed_data', 'top_weights', 'raw_SME_normalized'
varm: 'PCs'
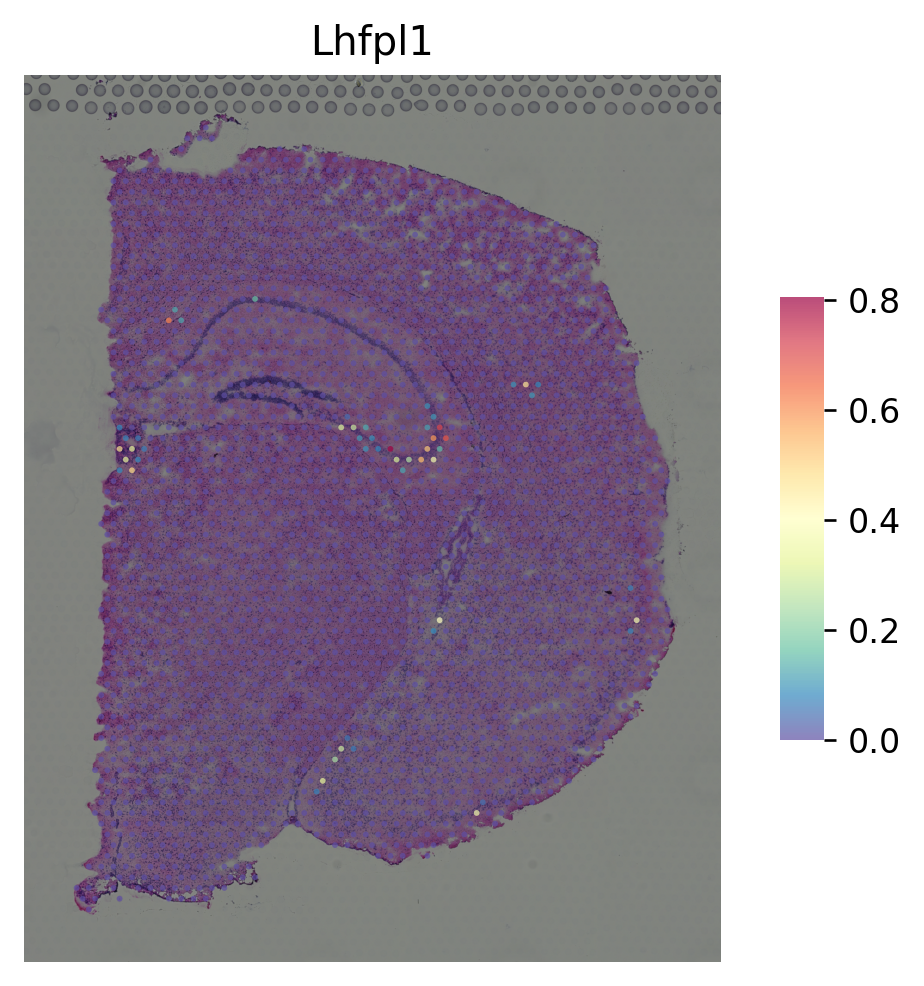
marker gene for DG¶
[12]:
i = "Pla2g2f"
st.pl.gene_plot(data_SME, gene_symbols=i, size=3)
[12]:
AnnData object with n_obs × n_vars = 2702 × 21949
obs: 'in_tissue', 'array_row', 'array_col', 'imagecol', 'imagerow', 'tile_path'
var: 'gene_ids', 'feature_types', 'genome', 'n_cells'
uns: 'spatial', 'log1p', 'pca', 'gene_expression_correlation', 'physical_distance', 'morphological_distance', 'weights_matrix_all', 'weights_matrix_pd_gd', 'weights_matrix_pd_md', 'weights_matrix_gd_md'
obsm: 'spatial', 'X_tile_feature', 'X_morphology', 'X_pca', 'imputed_data', 'top_weights', 'raw_SME_normalized'
varm: 'PCs'